Ziua Internațională a Studiilor Clinice 2026: despre studiile „First-in-Human” (Primele pe Om)


Întrebări și Răspunsuri cu John Bothmer, Clinical Translation Medicine Fellow la Angelini Pharma
Pentru a explora complexitatea științifică, de reglementare și operațională din spatele studiilor First-in-Human (primele pe om), am stat de vorbă cu John Bothmer, Clinical Translation Medicine Fellow în cadrul departamentului Global Clinical Development & Clinical Operations la Angelini Pharma, despre rolul acestor studii în dezvoltarea clinică și măsurile de siguranță care sprijină tranziția de la cercetarea preclinică la testarea pe oameni.
De la dovezile preclinice la studiile pe oameni: ce face ca un potențial medicament să fie gata pentru un studiu First-in-Human?
Înainte ca un potențial medicament să fie testat pe oameni, acesta trebuie să treacă printr-un proces riguros de pregătire care implică mai multe domenii interconectate.
În primul rând, trebuie să existe o justificare științifică clară: de ce a fost aleasă această țintă, de ce acest compus și de ce ar putea beneficia acest tip de pacienți. Această justificare trebuie să se bazeze pe date non-clinice care să confirme că substanța se comportă conform așteptărilor, precum și pe un program detaliat de siguranță și toxicologie, care să identifice riscurile potențiale, să stabilească o doză inițială sigură pentru prima administrare la oameni și să determine expunerea maximă care nu ar trebui să fie depășită.
De asemenea, calitatea joacă un rol esențial: produsul care urmează să fie investigat trebuie să respecte standarde stricte de fabricație pentru a garanta consistența și fiabilitatea. În plus, este necesar să existe aprobări de reglementare și etice înainte de a putea fi înscris vreun participant în studiu.
Este un proces solicitant — și pe bună dreptate. Tranziția de la laborator la testarea pe oameni reprezintă unul dintre cele mai importante etape în dezvoltarea unui medicament.
Cum este gestionată, monitorizată și revizuită siguranța în timpul unui studiu First-in-Human?
Siguranța în cadrul unui studiu FIH este gestionată proactiv — este integrată în design-ul studiului încă de la început.
Studiul urmează o abordare prudentă, cu escaladarea treptată a dozei: se începe cu doza maximă recomandată pentru prima administrare (Maximum Recommended Starting Dose) și se crește treptat doar după o analiză aprofundată a siguranței pentru fiecare nivel. În plus, un mic grup de participanți, numiți santinele, primește inițial doza, înainte ca aceasta să fie administrată întregului grup, pentru a asigura un nivel suplimentar de protecție.
Fiecare decizie de escaladare este revizuită de un Comitet Independent de Revizuire a Siguranței (Safety Review Committee), care evaluează toate datele disponibile în raport cu regulile prestabilite de oprire. Escaladarea nu este niciodată automată — este necesară o aprobare explicită. Dacă este atins un prag definit în protocolul de studiu, procesul este oprit.
Toate acestea au loc într-un mediu clinic controlat, cu o monitorizare continuă. Protecția participanților rămâne prioritatea centrală pe tot parcursul studiului.
Cine participă la studiile First-in-Human și cum sunt informați și protejați participanții?
Majoritatea studiilor FIH implică subiecți sănătoși – persoane care nu suferă de afecțiunea vizată și care aleg să contribuie la cercetările medicale din fazele incipiente. În unele cazuri, cum ar fi anumite programe din oncologie, unde profilul de risc al compusului îl face nepotrivit pentru subiecți sănătoși, pot participa în schimb pacienți.
Indiferent de cine participă, măsurile de protecție sunt aceleași. Studiul trebuie aprobat de un comitet de etică independent, iar fiecare participant trebuie să își dea consimțământul informat. Acest lucru înseamnă că aceștia primesc informații clare și oneste despre ce implică studiul, care sunt riscurile și că au libertatea de a se retrage oricând.
Acest proces nu este doar o formalitate. Este fundamentul etic al cercetării clinice.
De la prima administrare la oameni până la aprobarea de reglementare: care sunt următorii pași și de ce poate dura ani întregi?
Un studiu FIH este doar punctul de plecare, nu destinația finală. Rezultatele sale — intervalul de doze sigure, modul în care substanța se comportă în corpul uman și primele semne de activitate biologică — sunt utilizate direct în faza 2. Aici, medicamentul este studiat pentru prima dată la pacienți, iar eficacitatea terapeutică este testată, împreună cu identificarea celei mai potrivite doze.
Dacă rezultatele sunt promițătoare, programul trece în Faza 3: studii clinice de amploare, randomizate, desfășurate la nivel global, care implică, în funcție de indicație, sute sau chiar mii de participanți. Aceste studii sunt concepute pentru a confirma eficacitatea și pentru a construi un profil de siguranță solid.

Cum funcționează un studiu First-in-Human (FIH)
Studiile First-in-Human (FIH) sunt, de obicei, realizate în medii clinice extrem de controlate — cum ar fi clinici specializate sau spitale — concepute pentru a asigura monitorizarea continuă a participanților și intervenții rapide, dacă este necesar.
În majoritatea cazurilor, aceste studii implică grupuri mici de voluntari sănătoși. Totuși, atunci când medicamentul investigat prezintă un nivel mai ridicat de risc — de exemplu, în cazul anumitor tratamente oncologice unde compusul poate fi prea toxic pentru subiecți sănătoși — studiul poate fi realizat direct pe pacienți.
Un studiu FIH începe, de regulă, cu o fază de administrare unică a dozelor (Single Ascending Dose – SAD). Participanții primesc o singură doză a medicamentului investigat, începând cu o doză mică, stabilită pe baza dovezilor preclinice și definită ca doza maximă recomandată pentru prima administrare (Maximum Recommended Starting Dose – MRSD). După fiecare grup de participanți, datele privind siguranța și tolerabilitatea sunt analizate cu atenție înainte de a se decide creșterea dozei pentru următorul grup. Escaladarea dozei este oprită în momentul în care sunt atinse pragurile predefinite pentru expunere sau toxicitate.
Pentru a se reduce și mai mult riscurile, multe studii FIH includ subiecți santinele — de obicei, un singur participant care primește medicamentul investigat și un altul care primește placebo — înainte ca restul grupului să primească tratamentul. Această abordare permite evaluarea timpurie a siguranței înainte de expunerea întregului grup.
Atunci când se consideră că administrarea repetată a medicamentului este relevantă pentru utilizarea terapeutică viitoare, studiul poate progresa la o fază de administrare multiplă a dozelor (Multiple Ascending Dose – MAD). În această etapă, participanții primesc doze multiple pe o perioadă de timp definită — de exemplu, de două ori pe zi, timp de șapte zile. Și în această fază se utilizează o abordare prudentă de creștere treptată a dozelor, însoțită de o revizuire continuă a siguranței.
Deciziile privind escaladarea dozei, atât în faza SAD, cât și în faza MAD, sunt evaluate de un Comitet de Revizuire a Siguranței (Safety Review Committee – SRC), care analizează datele clinice, farmacocinetice și de siguranță în funcție de regulile prestabilite de oprire, incluse în protocolul studiului. Escaladarea dozei nu este automată: aceasta necesită o aprobare explicită din partea SRC, pe baza criteriilor definite anterior.
Deși obiectivul principal al acestor studii este evaluarea siguranței și tolerabilității, ele oferă, de asemenea, primele date despre farmacocinetica (modul în care medicamentul este procesat de organism) și despre efectele farmacodinamice timpurii (de exemplu, prin măsurarea biomarkerilor). Aceste informații sunt esențiale pentru a înțelege cum se comportă compusul investigat în organismul uman și pentru a ghida deciziile viitoare de dezvoltare clinică.
În concluzie, studiile FIH urmează o abordare progresivă și prudentă de creștere a dozelor, concepută pentru a asigura o evaluare continuă a siguranței pe tot parcursul studiului.

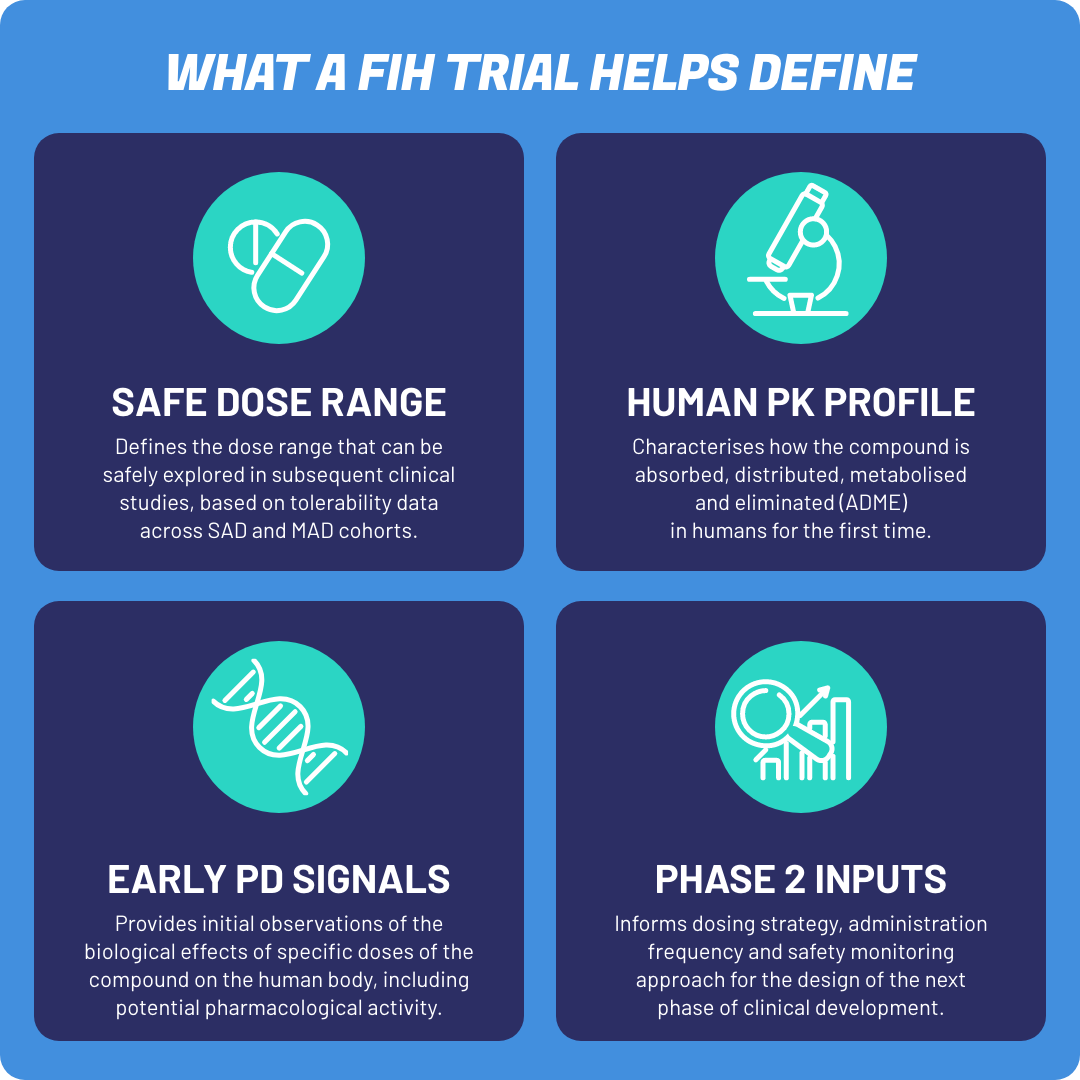
Importanța studiilor First-in-Human (FIH) în dezvoltarea clinică
Studiile First-in-Human oferă prima oportunitate de a transforma dovezile preclinice în cunoștințe clinice timpurii.
Datele generate în această etapă ajută la definirea intervalului de doze care poate fi explorat în studiile ulterioare și contribuie la înțelegerea relației expunere–răspuns, a comportamentului farmacocinetic și a aspectelor de siguranță asociate cu compusul investigat.
În mod important, studiile FIH furnizează, de asemenea, informații esențiale pentru proiectarea studiilor de fază 2, inclusiv decizii legate de schemele de dozare, frecvența administrării și strategiile de monitorizare a siguranței.
Deși aceste studii implică grupuri relativ mici de participanți, complexitatea lor științifică și operațională este considerabilă. Fiecare aspect al procesului — de la selecția dozei și strategia de escaladare până la monitorizarea participanților și evaluarea siguranței — este conceput pentru a sprijini luarea deciziilor informate, menținând în același timp protecția participanților ca prioritate centrală.
Din acest motiv, studiile First-in-Human nu reprezintă doar o etapă procedurală în dezvoltarea medicamentelor. Ele constituie un moment-cheie în transpunerea cercetării științifice în investigație clinică și în stabilirea fundamentelor pentru etapele următoare ale dezvoltării.
Deși relativ reduse ca amploare, studiile First-in-Human generează mai multe informații esențiale care ghidează dezvoltarea clinică ulterioară.